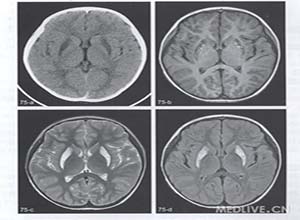
线粒体肌病(mitochondrial myopathy)是指因遗传基因的缺陷导致线粒体的结构和功能异常,导致细胞呼吸链及能量代谢障碍的一组多系统疾病。伴有中枢神经系统症状者称线粒体脑肌病。 此病于1962年由Luft首次采用改良Gomori Trichrome染色(MGT)发现肌纤维中有破碎红纤维(或不整红边纤维)(ragged red fiber,RRF),并诊断首例线粒体肌病,继而发现此类线粒体疾病也可同时累及中枢神经系统,引起多种线粒体脑肌病(mitochondrial encephalomyopathy)。本病为一组临床综合征。
无特定人群
0.002%
无力 糖尿 水肿 耳聋 肾功能衰竭 神经性耳聋 复视 视野缺损 麻痹 眼肌麻痹
无传染性
65%
1-3年
根据不同医院,收费标准不一致,市三甲医院约(5000――10000元)
肌电图
药物治疗 支持治疗
近年来研究已经发现某些疾病与mtDNA碱基替换突变有关。如Lerbe氏遗传性视神经病(Lerbe’s hereditary opticneuropathy)是mtDNA第11778位G转为A面使NADH脱氢酶第四亚单位ND4的第340位精氨酸残基被组氨酸残基取代,还有多个位点的突变对本病的发生起作用。又如线粒体脑肌病(mitochondrial mypathy,encephalopathy)伴高乳酸血症(lcticacidosis)和卒中样发作(stroke-like episodes)患者和成年型糖尿病伴耳聋患者,其mtDNA发生tRNALeu基因第3242位A→G替换突变。