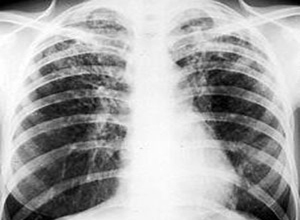
特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是一种不明原因的肺间质炎症性疾病,原因不明的肺泡纤维化和弥漫性肺间质纤维化均为其同义词。典型的IPF,主要表现为干咳、进行性呼吸困难,经数月或数年逐渐恶化,多在出现症状3~8年内进展至终末期呼吸衰竭或死亡。主要病理特点为肺间质和肺泡腔内纤维化和炎细胞浸润混合存在。尽管该病的发病机制还没有完全阐明,就其临床特征和病理足以说明这是一种特征性的疾病。IPF的治疗尚缺乏客观的、决定性的预后因素或治疗反应,皮质激素(以下简称激素)或免疫抑制剂、细胞毒药物仍是其主要的治疗药物,但不足30%的病人有治疗反应,且可表现毒副反应。
60岁以上老年男性多见,多有吸烟史
0.00025%
端坐呼吸 劳力性呼吸困难 干咳 一侧胸廓塌陷 脓痰 呼吸困难 呼吸衰竭 食欲不振 消瘦 紫绀
无传染性
70%
1-3个月
根据不同医院,收费标准不一致,市三甲医院约(5000-10000元)
呼气流速,肺功能检查,肺容积,肺显像,血氧饱和度
药物治疗 支持性治疗
许多疾病,尤其是引起免疫系统病变者,可引起肺纤维化。尽管已明确有许多可能的原因,但仍有半数肺纤维化患者的病因无法确定。这类原因不明的肺纤维化称为特发性肺纤维化(纤维化性肺泡炎,通常称间质性肺炎)。特发性一词意为原因不明。
特发性肺纤维化(idiopathicpulmonaryfibrosis,简称IPF),亦称隐源性纤维化肺泡炎或特发性间质性肺炎等。本病病因未明,弥散性肺间质纤维化局限于肺部。Hamman和Rich分别于1935年和1944年首先报道数例,均属急性型,进展急剧,在6周至半年内死亡,故又名Hamman-Rich综合症。本病多在40-50岁发病,男性稍多于女性,是遍及全世界的疾病,近10年来发病率及病死率皆升高,绝大多数为慢性型,发病年龄越高,病程越长。急性型罕见。近年多数学者认为系自身免疫性疾病,可能与遗传因素有关。